I have posted recently (e.g., 1, 2, 3) about a phytools function to mark changes on a plotted stochastic character map tree.
Well, one creative use of the latest update to this function might be, it occured to me, plotting arrows for changes of a particular type (say, decrease or increase in a discrete character trait on the tree, corresponding to 'up' or 'down' arrows, respectively). If a character tended to be lost but not regained, then, (for example, the number of digits) perhaps we would see losses but relatively few reaquisitions.
In the code below, I use a version of phytools that is not yet posted, but code for all the functions is already available on phytools.org.
Let's simulate data with a high rate of loss, & a low rate of reaquisition:
set.seed(872)
library(phytools)
Q<-matrix(c(-0.1,1,0,0,0,0,
0.1,-1.1,1,0,0,0,
0,0.1,-1.1,1,0,0,
0,0,0.1,-1.1,1,0,
0,0,0,0.1,-1.1,1,
0,0,0,0,0.1,-1),6,6,byrow=TRUE)
colnames(Q)<-rownames(Q)<-0:5
Q
## 0 1 2 3 4 5
## 0 -0.1 1.0 0.0 0.0 0.0 0
## 1 0.1 -1.1 1.0 0.0 0.0 0
## 2 0.0 0.1 -1.1 1.0 0.0 0
## 3 0.0 0.0 0.1 -1.1 1.0 0
## 4 0.0 0.0 0.0 0.1 -1.1 1
## 5 0.0 0.0 0.0 0.0 0.1 -1
tree<-sim.history(pbtree(n=50,scale=2),Q=Q,anc="5")
## Note - the rate of substitution from i->j should be given by Q[j,i].
## Done simulation(s).
colors<-setNames(grey(seq(1,0,-0.2)),0:5)
plotTree(tree,lwd=6,ftype="off",ylim=c(-1,Ntip(tree)))
plotSimmap(tree,colors=colors,ftype="off",add=TRUE,lwd=4,
ylim=c(-1,Ntip(tree)))
add.simmap.legend(x=0,y=0,prompt=FALSE,colors=colors,
vertical=FALSE)
obj<-markChanges(tree,plot=FALSE)
for(i in 1:nrow(obj)){
states<-as.numeric(strsplit(rownames(obj)[i],"->")[[1]])
up<-if(states[1]>states[2]) TRUE else FALSE
cex<-0.8
arrows(obj[i,1],obj[i,2]-0.8*mean(strheight(LETTERS)*cex),
obj[i,1],obj[i,2]+0.8*mean(strheight(LETTERS)*cex),
length=0.5*mean(strheight(LETTERS,units="inches")*cex),
lwd=3,col="red",ljoin=1,code=if(up) 1 else 2)
}
![plot of chunk unnamed-chunk-1]()
x<-getStates(tree,"tips") ## our data from simulation
sort(unique(x))
## [1] "0" "1" "2" "3" "4" "5"
OK, this is the real history. Now, let's imagine we don't know the history, but we suppose that loss & gain occur at different rates:
model<-matrix(c(0,1,0,0,0,0,
2,0,1,0,0,0,
0,2,0,1,0,0,
0,0,2,0,1,0,
0,0,0,2,0,1,
0,0,0,0,2,0),6,6)
rownames(model)<-colnames(model)<-0:5
model
## 0 1 2 3 4 5
## 0 0 2 0 0 0 0
## 1 1 0 2 0 0 0
## 2 0 1 0 2 0 0
## 3 0 0 1 0 2 0
## 4 0 0 0 1 0 2
## 5 0 0 0 0 1 0
x<-to.matrix(x,as.character(0:5))
smapsI<-make.simmap(tree,x,nsim=100,model=model)
## make.simmap is sampling character histories conditioned on the transition matrix
## Q =
## 0 1 2 3 4 5
## 0 -0.540027 0.540027 0.000000 0.000000 0.000000 0.000000
## 1 1.274384 -1.814411 0.540027 0.000000 0.000000 0.000000
## 2 0.000000 1.274384 -1.814411 0.540027 0.000000 0.000000
## 3 0.000000 0.000000 1.274384 -1.814411 0.540027 0.000000
## 4 0.000000 0.000000 0.000000 1.274384 -1.814411 0.540027
## 5 0.000000 0.000000 0.000000 0.000000 1.274384 -1.274384
## (estimated using likelihood);
## and (mean) root node prior probabilities
## pi =
## 0 1 2 3 4 5
## 0.1666667 0.1666667 0.1666667 0.1666667 0.1666667 0.1666667
## Done.
smapsI
## 100 phylogenetic trees with mapped discrete characters
obj<-summary(smapsI)
obj
## 100 trees with a mapped discrete character with states:
## 0, 1, 2, 3, 4, 5
##
## trees have 60.19 changes between states on average
##
## changes are of the following types:
## 0,1 0,2 0,3 0,4 0,5 1,0 1,2 1,3 1,4 1,5 2,0 2,1 2,3 2,4 2,5 3,0
## x->y 2.66 0 0 0 0 5.94 1.33 0 0 0 0 6.11 1.74 0 0 0
## 3,1 3,2 3,4 3,5 4,0 4,1 4,2 4,3 4,5 5,0 5,1 5,2 5,3 5,4
## x->y 0 9.54 4.25 0 0 0 0 10.41 6.56 0 0 0 0 11.65
##
## mean total time spent in each state is:
## 0 1 2 3 4 5
## raw 5.1938181 4.5646540 3.83968729 5.9647057 10.4758124 9.2228060
## prop 0.1322879 0.1162629 0.09779781 0.1519226 0.2668216 0.2349072
## total
## raw 39.26148
## prop 1.00000
plot(obj,colors=colors,ylim=c(-1,Ntip(smapsI[[1]])),ftype="off")
add.simmap.legend(x=0,y=0,prompt=FALSE,colors=colors,
vertical=FALSE)
![plot of chunk unnamed-chunk-2]()
We can similarly plot the location of reconstructed changes on any of stochastic mapped trees, for instance:
plotTree(smapsI[[1]],lwd=6,ftype="off",ylim=c(-1,Ntip(smapsI[[1]])))
plotSimmap(smapsI[[1]],colors=colors,ftype="off",add=TRUE,lwd=4,
ylim=c(-1,Ntip(smapsI[[1]])))
add.simmap.legend(x=0,y=0,prompt=FALSE,colors=colors,
vertical=FALSE)
obj<-markChanges(smapsI[[1]],plot=FALSE)
for(i in 1:nrow(obj)){
states<-as.numeric(strsplit(rownames(obj)[i],"->")[[1]])
up<-if(states[1]>states[2]) TRUE else FALSE
cex<-0.8
arrows(obj[i,1],obj[i,2]-0.8*mean(strheight(LETTERS)*cex),
obj[i,1],obj[i,2]+0.8*mean(strheight(LETTERS)*cex),
length=0.5*mean(strheight(LETTERS,units="inches")*cex),
lwd=3,col="red",ljoin=1,code=if(up) 1 else 2)
}
![plot of chunk unnamed-chunk-3]()
In this case, showing a pattern that is broadly similar to the generating pattern.
Finally, if we (for instance), knew that the ancestral state should have a particular value a priori - for instance, in this case, "5", we code set a strong prior on the state at the root to be "5". E.g.:
prior<-setNames(c(rep(0,5),1),0:5)
prior
## 0 1 2 3 4 5
## 0 0 0 0 0 1
smapsII<-make.simmap(tree,x,nsim=100,model=model,pi=prior)
## make.simmap is sampling character histories conditioned on the transition matrix
## Q =
## 0 1 2 3 4 5
## 0 -0.540027 0.540027 0.000000 0.000000 0.000000 0.000000
## 1 1.274384 -1.814411 0.540027 0.000000 0.000000 0.000000
## 2 0.000000 1.274384 -1.814411 0.540027 0.000000 0.000000
## 3 0.000000 0.000000 1.274384 -1.814411 0.540027 0.000000
## 4 0.000000 0.000000 0.000000 1.274384 -1.814411 0.540027
## 5 0.000000 0.000000 0.000000 0.000000 1.274384 -1.274384
## (estimated using likelihood);
## and (mean) root node prior probabilities
## pi =
## 0 1 2 3 4 5
## 0 0 0 0 0 1
## Done.
obj<-summary(smapsII)
obj
## 100 trees with a mapped discrete character with states:
## 0, 1, 2, 3, 4, 5
##
## trees have 59.41 changes between states on average
##
## changes are of the following types:
## 0,1 0,2 0,3 0,4 0,5 1,0 1,2 1,3 1,4 1,5 2,0 2,1 2,3 2,4 2,5 3,0
## x->y 2.26 0 0 0 0 5.58 1.14 0 0 0 0 5.98 1.48 0 0 0
## 3,1 3,2 3,4 3,5 4,0 4,1 4,2 4,3 4,5 5,0 5,1 5,2 5,3 5,4
## x->y 0 9.47 3.55 0 0 0 0 10.96 5.03 0 0 0 0 13.96
##
## mean total time spent in each state is:
## 0 1 2 3 4 5
## raw 5.1728761 4.5440518 3.68522497 5.1720941 9.6365903 11.0506463
## prop 0.1317545 0.1157382 0.09386362 0.1317346 0.2454464 0.2814628
## total
## raw 39.26148
## prop 1.00000
plot(obj,colors=colors,ylim=c(-1,Ntip(smapsII[[1]])),ftype="off")
add.simmap.legend(x=0,y=0,prompt=FALSE,colors=colors,
vertical=FALSE)
![plot of chunk unnamed-chunk-4]()










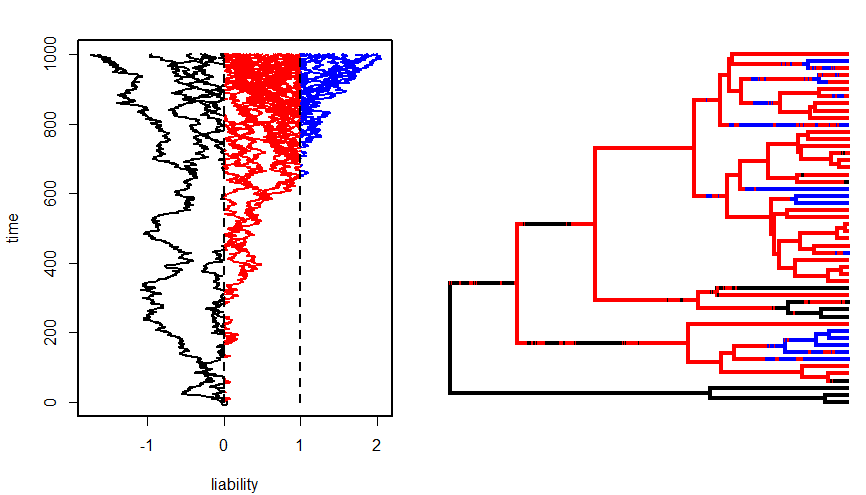 It is the final day of what was once the NESCent tutorial on Evolutionary Quantitative Genetics but has since become the NIMBioS
It is the final day of what was once the NESCent tutorial on Evolutionary Quantitative Genetics but has since become the NIMBioS 









































