The phytools function make.simmap performs stochastic mapping following Bollback (2006); however it does not implement a full hierarchical Bayesian model because stochastic maps are drawn conditional on the most probable value of the transition matrix, Q, found using maximum likelihood (given our data & tree - which are also assumed to be fixed). This is different from what (I understand) is implemented for morphological characters in the program SIMMAP, in which stochastic character histories & transition rates are jointly sampled, with a prior distribution on the parameters of the evolutionary model specified a priori by the user. Conditioning on the MLE(Q) (rather than integrating over the posterior distribution of Q) would seem to make what is done in make.simmap an empirical Bayes (EB) method.
From stochastic mapping we can compute posterior probability distributions on the number transitions between each pair of states; the total number of transitions; and the proportion of or total time spent in each state on the tree. (We can also get posterior probabilities of each state at internal nodes - i.e., ASRs - but these are boring &make.simmap does fine with this, so I won't focus on that here.)
I anticipated that make.simmap would have the following properties:
1. The point estimates of our variables of interest should be fine - i.e., unbiased and as good as the full hierarchical model.
2. The variance around those estimates computed from the posterior sample of stochastically mapped trees should be too small. This is because we have effectively ignored uncertainty in Q by fixing the transition matrix at its most likely value.
Finally, 3. the discrepancy in 2. should decline asymptotically for increasing tree size. This is because the more tips we have in our tree, the more information they will contain about the substitution process. In other words, the influence of ignoring uncertainty in Q should decrease as the information in our data about Q is increased.
These predictions are intuitive, but at least 1. & 2. are also typical of EB generally. Number 3. just seems sensible.
Unfortunately, since stochatic mapping is a computationally intensive Monte Carlo methods, testing these predictions is somewhat time consuming - and what I'll show now is only a very limited test. Basically, I simulated trees & data containing either 100 or 200 tips (and then 40 tips, see below) using pbtree and sim.history. The data are a binary character with states a and b, and the substitution model is symmetric - i.e., Qa,b = Qb,a. I wanted to see if our variables were estimated without (much) bias; and if (1-α)% CIs based on the posterior sample included the observed values (which we know from simulation, of course) the correct fraction of the time (e.g., 95% of the time for α = 0.05).
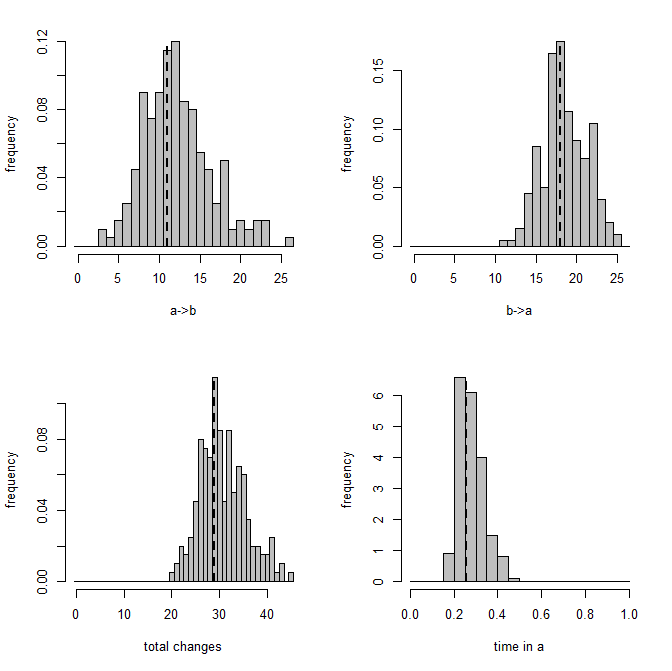
OK - I won't give all the code for simulation, but here is a figure showing a visualization of a single example result from one simulated 100-taxon tree & dataset. The panels give the transition rates between state, the total changes, or the relatively time spent in state a. (Since this last quantity is expressed as a proportion of the total time, the time spent in b is just 1 minus this.) The vertical dashed line is the value of each variable on the true tree.
This result was chosen at random, believe it or not (actually, it was the last of 100 simulations under the same conditions), but it happens to be a replicate in which make.simmap did really quite well.
Here is a table containing a summary of the first 10 of 100 simulations using 100-taxon trees, just to give the reader a sense of the data being collected. Hopefully the column headers are obvious.
> result[1:10,1:4]
a,b low.a,b mean.a,b high.a,b
1 8 2 5.820 11
2 7 3 5.750 10
3 9 10 19.155 30
4 15 11 15.660 20
5 5 5 8.780 12
6 4 2 7.030 14
7 13 9 11.940 16
8 8 11 17.420 25
9 5 3 6.810 12
10 7 10 18.650 28
> result[1:10,5:8]
b,a low.b,a mean.b,a high.b,a
1 16 12 15.070 19
2 10 7 10.410 14
3 21 16 25.510 34
4 5 2 5.940 12
5 5 3 5.875 10
6 17 13 17.265 22
7 8 5 8.295 12
8 18 10 17.490 24
9 12 7 11.005 15
10 18 9 18.150 26
> result[1:10,9:12]
N low.N mean.N high.N
1 24 15 20.890 28
2 17 11 16.160 22
3 30 32 44.665 60
4 20 15 21.600 29
5 10 11 14.655 20
6 21 16 24.295 32
7 21 14 20.235 27
8 26 25 34.910 45
9 17 13 17.815 25
10 25 27 36.800 48
> result[1:10,13:16]
time.a low.time.a mean.time.a high.time.a
1 0.229 0.196 0.244 0.310
2 0.296 0.225 0.289 0.361
3 0.335 0.271 0.392 0.541
4 0.762 0.651 0.743 0.805
5 0.364 0.305 0.394 0.467
6 0.182 0.173 0.230 0.322
7 0.485 0.410 0.456 0.505
8 0.320 0.333 0.441 0.549
9 0.201 0.209 0.284 0.397
10 0.260 0.294 0.423 0.565
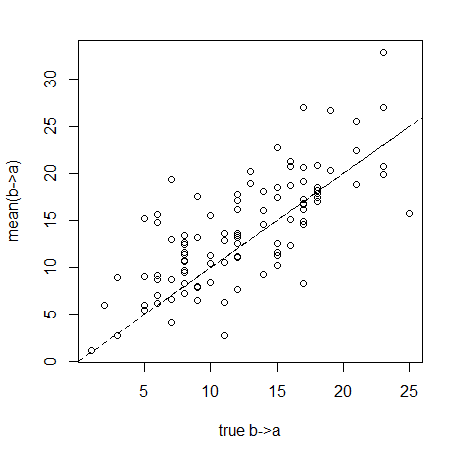
We can first check to see if the point estimates, obtained by averaging over stochastic maps for each simulation, give good estimates of the generating values for the variables that we are interested in. So, let's take the transitions from b to a as an example:
> plot(RR[,"b,a"],RR[,"mean.b,a"],xlab="true b->a", ylab="mean(b->a)")
> lines(c(0,max(RR[,"b,a"],RR[,"mean.b,a"])), c(0,max(RR[,"b,a"],RR[,"mean.b,a"])),lty="dashed")
We see that our point estimates track the known true number of transitions fairly well - so clearly we're not doing too bad with regard to bias. Let's quantify it across all the variables of interest:
> mean(RR[,"mean.a,b"]-RR[,"a,b"])
[1] 1.97765
> mean(RR[,"mean.b,a"]-RR[,"b,a"])
[1] 1.5159
> mean(RR[,"mean.N"]-RR[,"N"])
[1] 3.49355
> mean(RR[,"mean.time.a"]-RR[,"time.a"])
[1] 0.005261939
There looks to be a slight upward bias in the estimated number of substitutions - but this might just be due to the fact that our posterior sample is truncated at 0. (I.e., we might do better with the mode or median from the posterior sample instead of the arithmetic mean.) The time spent in
a (and thus also
b) seems to be estimated unbiasedly.
We can also ask, for instance, if the interval defined by [α/2, 1-α/2]% of the posterior sample includes the generating values (i.e., from our simulated tree) (1-α)% of the time. Setting α to 0.05:
> colMeans(on95)
a,b b,a N time.a
0.76 0.89 0.79 0.82
we see that indeed, and as expected, the variance on our variables is too small. Not way too small - what should be our 95% CI is actually our "76-89% CI" for these trees & data - but too small nonetheless.
Finally, prediction 3. Since I expect that the fact that our CIs are too small is due to fixing Q rather than integrating over uncertainty in Q (as we'd do in the full hierarchical model), I predicted that the variance of our parameters should asymptotically approach the true variance for more & more data. To "test" this, I simulated trees with 200 taxa and repeated the analyses above.
Here is one representative result, as before:
And let's check the average outcome across 100 simulations:
> colMeans(on95)
a,b b,a N time.a
0.85 0.87 0.77 0.86
To be honest, I was kind of surprised not to have found a larger effect here, so I decided to go in the other direction - and try simulating with quite small trees, say 40-taxa. First, here's a representative set of visualizations of the posterior densities/distributions for our focal variables in one randomly chosen simulation:
And here is our measure of the fraction of results on the (ostensible) 95% CI from the posterior:
> colMeans(on95)
a,b b,a N time.a
0.83 0.75 0.73 0.74
This result does seem to support premise 3., although, as for when we went from 100 to 200 tips, the effect of going from 100 to 40 is not especially large.
I should also be careful to note that this doesn't mean, of course, that we don't get much better parameter estimation from larger trees with more data - we do. It is just to say that convergence of our [α/2, 1-α/2]% to the true (1-α)% is flatter than I expected as sample size is increased, if it happens at all.
So, in summary - make.simmap does pretty well in point estimation. 95% CIs from the posterior sample will be too small - but not way too small, even for relatively small trees.
What's the way forward from here? Well, it would be nice to compare to SIMMAP, as I'm not aware of this kind of analysis having been done & published for morphological traits. In addition, there are steps that could be taken with make.simmap other than going to the full hierarchical model - for instance, instead of fixing Q at its MLE, I could use MLE(Q) to parameterize a prior probability distribution of the transition matrix. This would still be EB, just of a different flavor.
OK - that's all for now!